


2020-07-06 by Quick Biology Inc.
The CRISPR-Cas system is adapted from a naturally occurring genome editing system in bacteria and archaea, it is a prokaryotic immune system that confers resistance to foreign genetic elements. This CRISPR-associated system is highly expanded and developed by Dr. Jennifer Doudna in UC, Berkeley (https://doudnalab.org/), and Dr. Feng Zhang at Broad Institute (https://zlab.bio/). Currently, the CRISPR-Cas system offers the eukaryotic genome (DNA level) and epigenome editing (RNA level), which can be repurposed in gene activation or repression via CRISPR-Cas or variants fusion to other modifiers.
Although targeting DNA or RNA by the CRISPR-Cas system has great precision compared to traditional genome editing, transgenic systems, off-target editing is still a good concern, especially for therapeutic and clinical applications. In current Molecular Cell, Dr. Clement and colleagues briefly presented CRISPR technologies and particularly focused on the analysis of Next Generation Sequencing data for on-and off-target editing and CRISPR pooled screen strategies (ref1). This nice minireview summarized current tools, computational perspectives, and analytical challenges of CRISPR-Cas toolbox, off-target screening assays (Fig.1 to 3).
Figure 1: Overview of Strategies to Detect Editing at Known Loci Including Heteroduplex DNA Analysis, Loss of Binding Site Analysis, and Sequencing-Based Approaches. In heteroduplex DNA analysis (left panel), heteroduplex complexes between wild-type (non-edited) and nuclease-edited DNA strands (edited bases are shown in red) are formed by denaturing and annealing PCR amplicons generated from a bulk population of edited cells. Mismatches in heteroduplex DNA are detected and cleaved by enzymes such as T7E1 or Surveyor. Loss of binding site analysis (middle panel) relies on the ability of a PCR primer (green DNA sequence) to bind based on Watson-Crick complementarity or a transcription factor or restriction enzyme to identify its recognition sequence. Editing can be identified by binding site modification that results in the loss of PCR primer binding or loss of restriction enzyme mediated cleavage. In sequencing-based assays (right panel), Sanger sequencing or Next Generation Sequencing (NGS) are used to analyze a given site (Adapted from Ref 1).

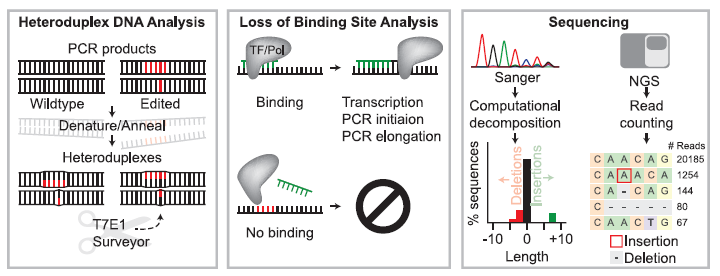
Figure 2: Overview of In Silico, In Vitro, and In Cellula Strategies to Nominate Off-Target Editing at Known or Unknown Loci. In silico approaches (left panel) utilize sequence homology to identify genomic loci (top strand) with similarity to the gRNA sequence (bottom strand) up to a particular number of mismatches. Other approaches use enzymatic modeling to predict gRNA binding specificity at putative off-target sites. Machine learning approaches have also been developed to identify off-target sites. In vitro approaches (middle panel) first extract DNA from cells, and in CIRCLE-seq, DNA is circularized. Next DNA is exposed to editing reagents and cleaved fragments can be selected and sequenced to identify off-target cleavage or subjected to whole-genome re-sequencing with identification of cleavage sites by identifying reads that start or end at the same base position. In cellula strategies (right panel) introduce CRISPR reagents into cells in the native cellular context. Cleavage events can be detected through a variety of methods such as ligation of known sequences to double-strand breaks, biochemical tagging with biotinylated primers, or immunoprecipitation of DNA repair factors recruited to sites of cleavage (Adapted from Ref 1).
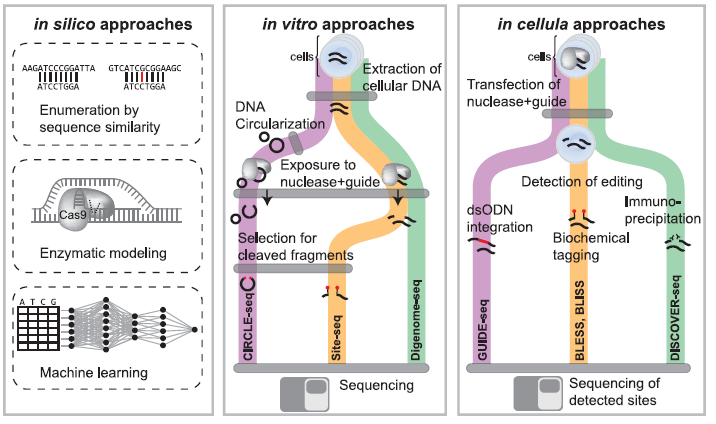
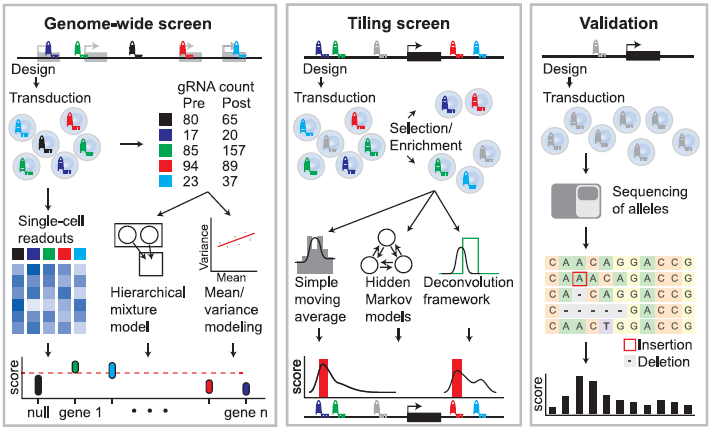
Figure 3: Overview of Analysis Strategies for Tiling and Gene-Targeted Pooled Screens followed by Screen Validation. Genome-wide screening approaches (left panel) utilize gene-targeted gRNA libraries in viral vectors. gRNA abundance is determined before and after phenotypic selection or enrichment. Scores are generated by comparing the relative gRNA abundance in pre-and post-selection populations, and identification of critical genes is performed using mean/variance modeling to address overdispersion or hierarchical mixture models to accommodate gRNA- and gene-specific variation. Alternately, single-cell readouts such as scRNA-seq can be applied to populations of cells to link phenotypes to specific perturbations. Tiling screens (middle panel) are performed by targeting gRNAs across a genomic interval. gRNA abundance is determined before and after phenotypic selection/enrichment. Scores are generated by comparing the relative gRNA abundance in pre-and post-selection populations. The effect of each gRNA can be computed using simple moving averages, hidden Markov models, or deconvolution frameworks. Pooled screen validation (right panel) often involves re-testing gRNAs in an arrayed format in bulk cell populations or individual clones using the techniques for measuring editing at known loci. For example, next-generation sequencing of an individual gRNA target site can be followed by computational analysis to identify generated alleles and calculate a per-base activity score (Adapted from Ref 1).
Quick Biology is an expert in providing NGS CRISPR screening services. Find More at Quick Biology.
Ref:
1. Clement, K., Hsu, J. Y., Canver, M. C., Joung, J. K. & Pinello, L. Technologies and Computational Analysis Strategies for CRISPR Applications. Mol. Cell 79, 11–29 (2020).