


2019-08-25 by Quick Biology Inc.
Methylation status of DNA and accessibility of chromatin structure are often correlated with gene expression in eukaryotic cells. Using whole-genome bisulfite sequencing (WGBS), researchers can profile DNA methylome, while Hi-C is a comprehensive technique to capture the conformation of genomes.
In recent Nature Methods, Bing Ren group at UCSD developed a joint profiling method called Methyl-HiC, which simultaneously collects information of DNA methylation and three-dimensional chromatin folding. The idea is simple, based on the premise that methylation status of DNA is preserved in chromosome conformation, authors began with a standard in situ Hi-C procedure, then followed by bisulfite conversion before library construction and next generation sequencing (Figure 1). Using mouse embryonic stem cells as test samples, Bing Ren group evaluated Methyl-HiC, that had a great agreement with separate WGBS or Hi-C profiling (Figure 2).
Figure 1: Schematic of Methyl-Hi C protocol, in which biotin-enriched DNA fragments from in situ Hi-C are bisulfite converted followed by paired-end sequencing. The short sequencing reads are mapped to the genome. (ref1)
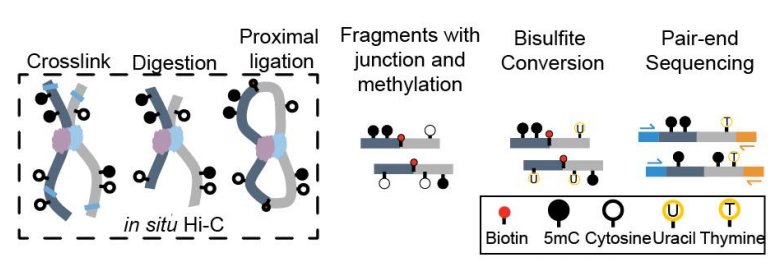
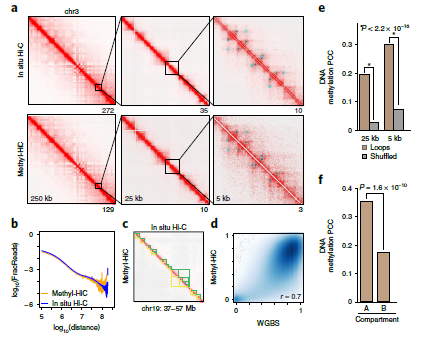
Figure 2: Profiling comparison between Methyl-HiC and Hi-C or WGBS. Methyl-HiC simultaneously profiles long-range chromatin interactions and DNA methylome in mESCs. (ref1)
Quick Biology is an expert in NGS library preparation. We are currently providing WGBS and Hi-C seq services. Are you ready to sequence your samples of interest? Find More at Quick Biology.
Reference:
- 1. Li, G. et al. Joint profiling of DNA methylation and chromatin architecture in single cells. Nat. Methods (2019). doi:10.1038/s41592-019-0502-z
- 2. Clark, S. J. et al. ScNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells e. Nat. Commun. 9, 1–9 (2018).