


2020-12-22 By Quick Biology
Stem cell therapy or regenerative medicine promotes the repair response of diseased, injured, or dysfunctional tissues using stem cells. The important biosafety for the use of human pluripotent stem cells is to make sure there are not any mutations, especially in genes associated with cancer that occurred during stem cell in vitro propagation.
In Cell Stem Cell, to clarify the prevalence of cancer-related mutations in naïve hPSCs, Stirparo and colleagues re-examined RNA-seq data from different cultures of Cr-S6EOS and other naïve cell lines (Ref1). By applying the established GATK pipeline for calling SNPs from RNA-seq data, and performing XenofilteR, a tool previously developed for the analysis of human xenografts in mice. They identified and removed reads that map with higher efficiency to the mouse than to the human reference genome (Fig.1). They observed that, that those mutations reported by Avior et al. 2019 were only present in co-cultures with mouse feeder cells (Fig. 2). The detection of multiple mutations in naïve stem cells by RNA-seq analysis is due to contamination with mouse sequences. Furthermore, they did another independent analysis using the metagenomic tool Sequence Expression AnaLyzer (SEAL) to classify human or mouse sequences and yielded similar results. After they filtered reads to remove sequences of mouse origin, the actual incidence of oncogenic polymorphisms arising in naïve pluripotent stem cell is close to zero.
Avior and colleagues recognized their unintentional absence of such reads filter, they retracted their 2019 paper (Cell Stem Cell 25, 456-461) and directed readers to their new Letter to the journal.
Figure 1: Analysis of Genomic Origin of SNPs Reported by Avior et al. 2019. (ref1)
(A) Numbers of cancer-associated SNPs from Avior et al. (2019) in cR-S6EOS samples cultured on mouse embryo fibroblast (MEF) feeder layers or on laminin (LN) detected by the GATK pipeline without variants hard-filtering step. (B) Integrative Genome Browser screenshot of selected cancer-associated SNPs from Avior et al. (2019) showing per base read coverage (0–100) in cR-S6EOS cultures on MEF or LN. Dotted lines highlight the SNP reported by Avior et al. (2019). Positions with alternative nucleotides are represented using different colors. (C) Average mapping percentage of total reads from cR-S6EOS(MEF) samples harboring the indicated SNPs reported by Avior et al. (2019) when aligned against human or mouse reference sequences. See also Tables S1 and S2. (D) Number of mismatches in reads as in (C) aligned against human or mouse reference sequences. (E) Boxplots of the number of mouse reads detected by XenofilteR in naive cell samples from cultures on MEF or LN. (F) Boxplots of the number of mouse reads identified by XenofilteR in naive and primed conditions across different datasets analyzed in Avior et al. (2019).
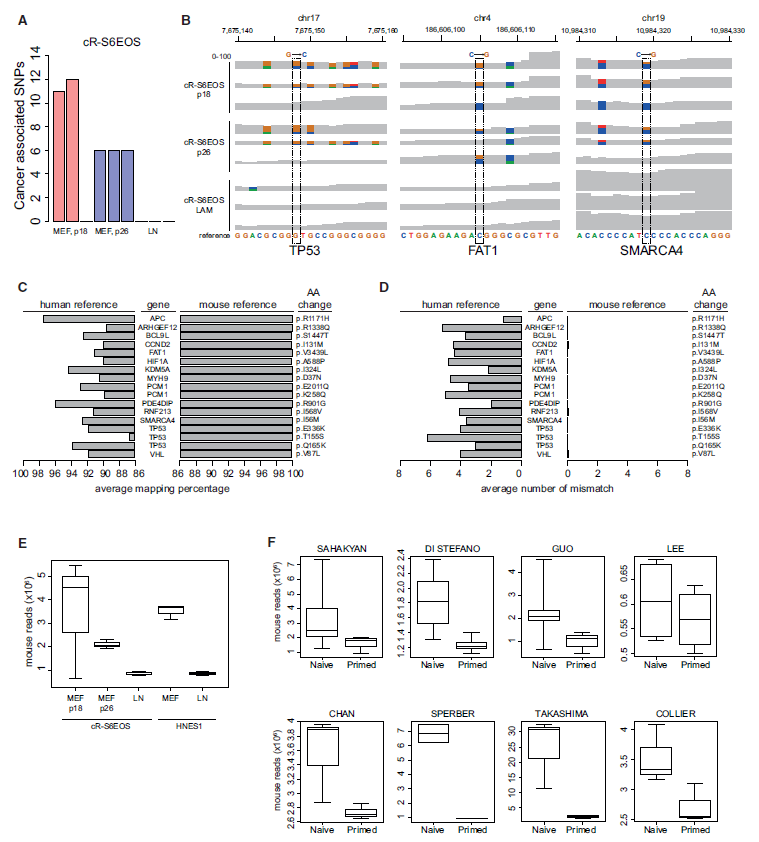
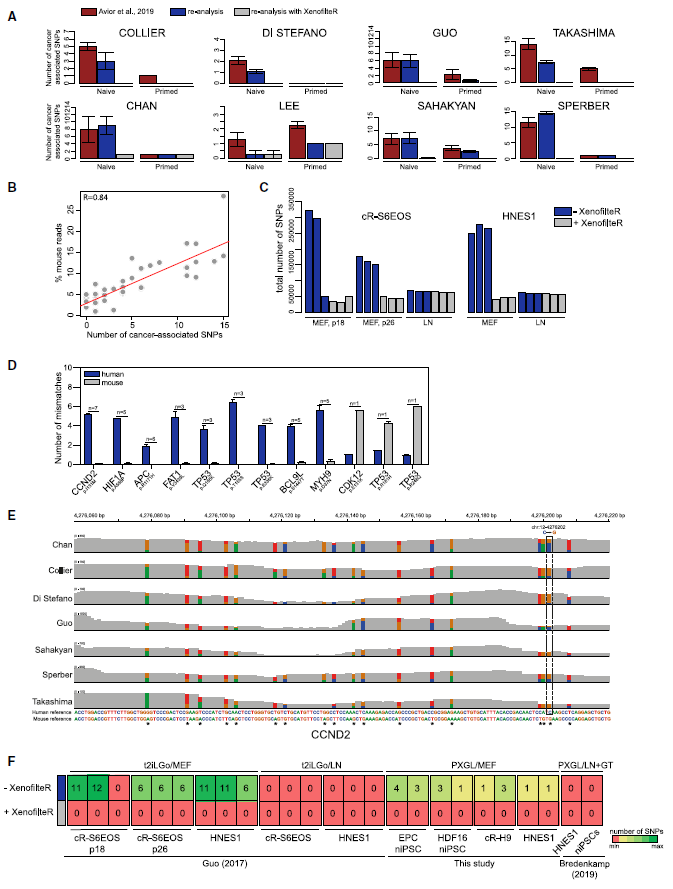
Figure 2: Elimination of Mouse Sequences Removes Cancer-Associated SNPs. (Ref1)
(A) Number of cancer-associated SNPs from Avior et al. (2019) in different datasets, as reported in Avior et al., (2019) (red), as detected in this study without XenofilteR (blue), and as detected after removal of mouse reads using XenofilteR (grey). Error bars, SEM. (B) Correlation between percentage of mouse reads and numbers of cancer-associated SNPs detected for all naive hPSCs in this study. (C) Total number of SNPs before and after removal of mouse reads in cR-S6EOS and HNES1 cultures on MEF or LN. (D) Numbers of mismatches in reads harboring the cancer-related mutation aligned against human or mouse reference sequences. Each bar represents average number of mismatches for all reads with SNPs reported by Avior et al. (2019) in naive hPSCs. n represents number of datasets with the indicated SNP. Error bars, SEM. (E) Integrative Genome Browser screenshot of CCND2 transcripts showing the SNP reported by Avior et al. (2019) in dashed box and nearby mismatches in reads across indicated human naive hPSC datasets. (F) Heatmap showing number of Avior et al. (2019) SNPs detected in human naive hPSCs cultured in t2ilGo¨ medium or PXGL medium on MEF or on LN with or without application of XenofilteR. Samples from Bredenkamp et al. (2019b) are pooled data from cultures on LN or Geltrex (GT).
“When using NGS to identify mutations and single nucleotide polymorphisms in hPSCs, it is vital to use tools that segregate human and murine sequences, as these could interfere with the correct interpretation of results” as said by Avio et al. in their clarification. Therefore, it is crucial to avoid read contaminations when doing RNA-seq, Quick Biology can assistant you with filtering RNA-seq reads from different species. Find More at Quick Biology.
See resource:
1. Stirparo, G. G. et al. Matters Arising Cancer-Related Mutations Are Not Enriched in Naive Human Pluripotent Stem Cells Cancer-Related Mutations Are Not Enriched in Naive Human Pluripotent Stem Cells. Stem Cell 1–6 (2021). doi:10.1016/j.stem.2020.11.014